УТВЕРЖДЕНА
Приказом Председателя РГУ «Комитет медицинского и фармацевтического контроля Министерства здравоохранения Республики Казахстан» от «23» апреля 2025 года
№ N085306
Общая характеристика лекарственного препарата
1. Наименование лекарственного препарата
Козэнтикс, 150 мг/мл, раствор для подкожного введения
2. Качественный и количественный состав
2.1 Общее описание
Секукинумаб*
2.2 Качественный и количественный состав
Один предварительно заполненный шприц содержит
Секукинумаб, 150 мг
*Секукинумаб представляет собой рекомбинантное полностью человеческое моноклональное антитело, выращенные в клетках яичников китайского хомячка (CHO).
Полный список вспомогательных веществ см. в пункте 6.1.
3. Лекарственная форма
Раствор для подкожного введения.
Прозрачный или опалесцирующий раствор от бесцветного до светло-желтого цвета.
4. Клинические данные
Лечение бляшечного псориаза среднетяжелой и тяжелой степени у взрослых пациентов, которым показана системная терапия
Бляшечный псориаз у детей
Лечение умеренной или тяжелой степени бляшечного псориаза у детей и подростков старше 6 лет, которые являются кандидатами на системную терапию.
Псориатический артрит
Лечение активного псориатического артрита в монотерапии или в комбинации с метотрексатом у взрослых пациентов при недостаточном ответе на предшествующую терапию болезнь модифицирующими антиревматическими препаратами (БМАРП) (см. раздел 5.1).
Аксиальный спондилоартрит
Анкилозирующий спондилит (рентгенологический аксиальный спондилоартрит)
Лечение активного анкилозирующего спондилита у взрослых пациентов при недостаточном ответе на стандартную терапию.
Лечение активного нерентгенологического аксиального спондилоартрита с объективными симптомами воспалительного процесса, на что указывает повышенное содержание С-реактивного белка (СРБ) и (или) магнитно-резонансная томография (МРТ) у взрослых без адекватного ответа на лечение нестероидными противовоспалительными лекарственными средствами (НПВС).
Ювенильный идиопатический артрит (ЮИА)
Энтезит-ассоциированный артрит (ЭАА)
Козэнтикс в режиме монотерапии или в комбинации с метотрексатом (МТХ) показан для лечения активного энтезитассоциированного артрита у пациентов в возрасте 6 лет и старше, у которых не был достигнут адекватный ответ на традиционную терапию или возникла ее непереносимость (см. раздел 5.1)
Козэнтикс в режиме монотерапии или в комбинации с метотрексатом (МТХ) показан для лечения активного ювенильного псориатического артрита у пациентов в возрасте 6 лет и старше, у которых не был достигнут адекватный ответ на традиционную терапию или возникла ее непереносимость (см. раздел 5.1).
Препарат Козэнтикс предназначен для использования под руководством и наблюдением врача, имеющего опыт в диагностике и лечении состояний, при которых показано применение препарата Козэнтикс.
Рекомендуемая доза составляет 300 мг секукинумаба в виде подкожной инъекции с начальной дозой на 0, 1, 2, 3 и 4 неделях с последующим ежемесячным введением поддерживающей дозы. Основываясь на клиническом ответе, поддерживающая доза 300 мг каждые 2 недели может обеспечить дополнительный положительный эффект для пациентов с массой тела 90 кг или выше. Каждая доза в 300 мг вводится в виде двух подкожных инъекций по 150 мг.
Бляшечный псориаз у детей и подростков старше 6 лет
Рекомендуемая доза рассчитывается по массе тела (таблица 1) и вводится подкожно с начальной дозой на 0, 1, 2, 3 и 4 неделю, с последующим ежемесячным введением поддерживающей дозы.
Каждая доза в 75 мг вводится в виде одной подкожной инъекции объемом 75 мг. Каждая доза в 150 мг вводится в виде одной подкожной инъекции объемом 150 мг. Каждая доза в 300 мг вводится в виде двух подкожных инъекций по 150 мг.
Таблица 1. Рекомендованные дозы при бляшечном псориазе у детей

Раствор для инъекций в предварительно заполненной шприц-ручке и предварительно заполненном шприце в автоинжекторе (ручке) 150 мг и 300 мг не показан для введения детям с массой тела <50 кг. Препарат Козэнтикс может быть доступен в других дозировках и / или формах выпуска в зависимости от индивидуальных потребностей в лечении.
Псориатический артрит
Пациентам с сопутствующим бляшечным псориазом средней и тяжелой степени следует придерживаться рекомендаций по бляшечном псориазу для взрослых.
У пациентов с неадекватным ответом на терапию ингибиторами ФНО-α рекомендуемая доза составляет 300 мг в виде подкожной инъекции в качестве начальной дозы на 0, 1, 2, 3 и 4 неделе с последующим ежемесячным введением в качестве поддерживающей дозы. Каждая доза в 300 мг вводится в виде двух подкожных инъекций по 150 мг.
У других пациентов рекомендуемая доза составляет 150 мг в виде подкожной инъекции в качестве начальной дозы на 0, 1, 2, 3 и 4 неделе, с ежемесячным поддерживающим дозированием. На основании клинического ответа доза может быть увеличена до 300 м
Аксиальный спондилоартрит
Анкилозирующий спондилит (рентгенологический аксиальный спондилоартрит)
Рекомендуемая доза составляет 150 мг в виде подкожной инъекции в качестве начальной дозы на 0, 1, 2, 3 и 4 неделе с последующим ежемесячным приемом поддерживающей дозы. На основании клинического ответа доза может быть увеличена до 300 мг. Каждая доза в 300 мг вводится в виде двух подкожных инъекций по 150 мг.
Нерентгенологический аксиальный спондилоартрит (нр-аксСПА)
Рекомендуемая доза составляет 150 мг в виде подкожной инъекции в качестве начальной дозы на 0, 1, 2, 3 и 4 неделе, с последующим ежемесячным приемом поддерживающей дозы
Ювенильный идиопатический артрит (ЮИА)
Энтезит-ассоциированный артрит (ЭАА) и ювенильный псориатический артрит (ЮПА)
Рекомендуемая доза рассчитывается по массе тела (таблица 2) и вводится подкожно в 0, 1, 2, 3 и 4 неделю с ежемесячной коррекцией дозировки. Каждая доза в 75 мг вводится в виде одной подкожной инъекции объемом 75 мг. Каждая доза в 150 мг вводится в виде одной подкожной инъекции объемом 150 мг.
Таблица 2. Рекомендуемые дозы при ювенильном идиопатическом артрите

Козэнтикс может быть доступен в других дозировках и (или) лекарственных формах в зависимости от индивидуальных терапевтических потребностей.
По всем указанным выше показаниям доступные данные указывают на то, что клинический ответ обычно достигается в течение 16 недель лечения.
Следует рассмотреть возможность прекращения лечения пациентам, у которых не было ответа к 16 неделе лечения. У некоторых пациентов с первоначально частичным ответом может впоследствии улучшиться состояние после продолжающегося лечения спустя 16 недель
Коррекция дозы не требуется (см. раздел 5.2).
Пациенты с печеночной и почечной недостаточностью
Козэнтикс не изучался у пациентов данной популяции. Рекомендации о дозировке отсутствуют.
Дети
Безопасность и эффективность препарата Козэнтикс у детей с бляшечным псориазом и при ювенильном идиопатическом артрите (ЮИА) категорий ЭАА и ЮПА в возрасте до 6 лет не изучены.
Безопасность и эффективность препарата Козэнтикс у детей в возрасте младше 18 лет при других показаниях пока не установлены. Данные отсутствуют.
Козэнтикс следует вводить подкожно. По возможности следует избегать инъекций на участках кожи, пораженных псориазом. Раствор/ автоинжектор (ручку) запрещается встряхивать.
После надлежащего обучения технике введения препарата пациенты могут самостоятельно вводить препарат Козэнтикс, если такая необходимость подтверждена врачом. Тем не менее, врач должен обеспечить соответствующее наблюдение за пациентами. Пациенты или лица, осуществляющие уход, должны получить информацию о том, как вводить препарат Козэнтикс в полном объеме в соответствии с инструкциями, приведенными в листке-вкладыше. Подробная инструкция по применению приведена в листке-вкладыше.
- Гиперчувствительность к действующему веществу или к любому из вспомогательных веществ, перечисленных в разделе 6.1
- Клинически значимые инфекции в стадии обострения (например, активный туберкулез, см. раздел 4.4)
Чтобы улучшить прослеживаемость биологических лекарственных средств, следует точно записать название и номер серии вводимого препарата.
Инфекции
Секукинумаб может увеличивать риск развития инфекций. В постмаркетинговых исследованиях у пациентов, получавших секукинумаб, отмечалось развитие серьезных инфекций. Следует соблюдать осторожность при назначении секукинумаба пациентам с хроническими инфекциями или рецидивирующими инфекциями в анамнезе.
Пациенты должны быть проинструктированы о необходимости обращения к врачу при появлении признаков и симптомов, указывающих на развитие инфекции. Если у пациента развивается серьезная инфекция, то необходимо обеспечить тщательное наблюдение за пациентом и отмену введения секукинумаба до устранения симптомов инфекции.
В клинических исследованиях у пациентов, получающих секукинумаб, отмечалось развитие инфекций (см. раздел 4.8). Большинство из них были инфекциями верхних дыхательных путей легкой и средней степеней тяжести, например, назофарингит, и не требовали прекращения лечения.
Связанные с механизмом действия секукинумаба несерьезные кожнослизистые кандидозы чаще отмечались у пациентов, получавших секукинумаб, чем у пациентов, получавших плацебо, в клинических исследованиях псориаза (3,55 на 100 пациенто-лет для 300 мг секукинумаба по сравнению с 1,00 на 100 пациенто-лет в группе плацебо) (см. раздел 4.8).
В клинических исследованиях не сообщалось о повышенной чувствительности к туберкулезу. Тем не менее, секукинумаб не следует назначать пациентам с активной формой туберкулеза. Необходимо назначить противотуберкулезную терапию пациентам с латентной формой туберкулеза до начала приема секукинумаба.
Воспалительные заболевания кишечника (включая болезнь Крона и язвенный колит)
Сообщалось о новых случаях или эпизодах обострения воспалительного заболевания кишечника при применении секукинумаба. Секукинумаб не рекомендуется пациентам с воспалительными заболеваниями кишечника. Если у пациента появляются признаки и симптомы воспалительного заболевания кишечника или наблюдается обострение ранее существовавшего воспалительного заболевания кишечника, прием секукинумаба следует прекратить и начать соответствующее медицинское лечение.
Реакции гиперчувствительности
У пациентов, получающих секукинумаб, отмечались редкие случаи анафилактических реакций и ангионевротического отека. При возникновении анафилактической реакции, ангионевротического отека или других серьезных аллергических реакций необходимо немедленно прекратить прием секукинумаба и инициировать соответствующую терапию.
Чувствительность к латексу
Съемная крышка предварительно заполненного шприца и шприца в автоинжекторе (ручке) с препаратом Козэнтикс содержит производные натурального каучукового латекса. На текущий момент натуральный каучуковый латекс в съемной крышке не обнаружен. Тем не менее, использование предварительно заполненных шприцов и шприцов в автоинжекторе (ручке) с препаратом Козэнтикс у пациентов с аллергией на латекс не было изучено, поэтому существует потенциальный риск возникновения реакций гиперчувствительности, который не может быть полностью исключен.
Вакцинация
Живые вакцины не следует назначать одновременно с секукинумабом.
Пациенты, получающие секукинумаб, могут одновременно получать инактивированные или неживые вакцины. В исследовании равное соотношение здоровых добровольцев, получавших 150 мг секукинумаба, и пациентов, получавших плацебо, наблюдался адекватный иммунный ответ после вакцинации менингококковой и инактивированной вакцины против гриппа, что проявлялось в виде 4-кратного увеличения титров антител к менингококковой вакцине и вакцине против гриппа. Эти данные свидетельствуют о том, что секукинумаб не подавляет гуморальный иммунный ответ на менингококковые вакцины или вакцины против гриппа.
Рекомендуется, чтобы все пациенты детского возраста до начала приема препарата Козэнтикс получили соответствующие возрасту прививки в соответствии с рекомендациями по иммунизации.
Одновременное применение иммуносупрессивной терапии
В исследованиях псориаза безопасность и эффективность секукинумаба в сочетании с иммунодепрессантами, в том числе биологическими, или фототерапией не были оценены. В клинических исследованиях пациентам, страдающим артритами (в том числе пациентам с псориатическим артритом и анкилозирующим спондилитом), секукинумаб вводили одновременно с метотрексатом (MTX), сульфасалазином и/или кортикостероидами. Следует соблюдать осторожность при рассмотрении вопроса об одновременном использовании других иммунодепрессантов и секукинумаба (см. также раздел 4.5).
Реактивация гепатита B
Реактивация вируса гепатита B может произойти у пациентов, проходящих лечение секукинумабом. В соответствии с клиническими рекомендациями по иммунодепрессантам, перед началом лечения секукинумабом следует рассмотреть возможность тестирования пациентов на инфекцию вируса гепатита B. Пациентов с положительным результатом серологического исследования на вирус гепатита B следует контролировать на предмет клинических и лабораторных признаков реактивации вируса гепатита B во время лечения секукинумабом. Если реактивация вируса гепатита B происходит во время лечения секукинумабом, следует рассмотреть возможность прекращения лечения, и пациентов следует лечить в соответствии с клиническими рекомендациями.
Секукинумаб не следует назначать одновременно с живыми вакцинами (см. также раздел 4.4).
В исследовании с участием пациентов с бляшечным псориазом не выявлено взаимодействия между секукинумабом и мидазоламом (субстратом CYP3A4). Не выявлено лекарственного взаимодействия секукинумаба с метотрексатом (МТХ) и/или кортикостероидами в ходе исследований артрита (включая пациентов с псориатическим артритом и аксиальным спондилоартритом).
Женщины детородного возраста должны использовать эффективные методы контрацепции во время лечения и в течение, по крайней мере, 20 недель после лечения.
Беременность
Достоверные данные об использовании секукинумаба у беременных женщин отсутствуют.
Исследования на животных не указывают на прямое или косвенное негативное токсическое влияние на репродуктивную функцию (см. раздел 5.3). В качестве меры предосторожности желательно избегать использования препарата Козэнтикс во время беременности.
Кормление грудью
Не установлено, выделяется ли секукинумаб в материнское молоко.
Иммуноглобулины выделяются в материнское молоко, но не известно, абсорбируется ли секукинумаб системно после приема.
Из-за возможности нежелательных реакций на секукинумаб у вскармливаемых грудным молоком младенцев целесообразно рассмотреть возможность прекращения грудного вскармливания во время лечения и до 20 недель после лечения или прекращения лечения препаратом Козэнтикс, принимая во внимание соотношение пользы кормления грудью ребенка и пользы лечения препаратом Козэнтикс для женщины.
Фертильность
Эффект секукинумаба на фертильность человека не был оценен.
Исследования на животных не подтверждают прямые или косвенные вредные последствия в отношении фертильности.
Козэнтикс не имеет или имеет незначительное влияние на способность управлять транспортными средствами и механизмами.
К наиболее часто упоминаемым нежелательным лекарственным реакциям (НЛР) относятся инфекции верхних дыхательных путей (17,7%) (чаще всего - назофарингит, ринит).
НЛР, выявленные в ходе клинических исследований, а также НЛР по данным пострегистрационного применения (таблица 3), перечислены в соответствии с системно-органными классами Медицинского словаря терминологии регулятивной деятельности (MedDRA). В пределах каждого системно-органного класса НЛР распределены по частоте возникновения в порядке уменьшения частоты. В каждой группе по частоте нежелательные реакции представлены в порядке убывания их степени тяжести. Кроме того, соответствующая категория частоты для каждой нежелательной лекарственной реакции основана на следующем порядке: очень часто (≥1/10); часто (≥1/100 до <1/10); нечасто (≥1/1000 до <1/100); редко (от ≥ 1/10 000 до <1/1000); очень редко (<1/10 000); неизвестно (невозможно оценить по имеющимся данным).
В ходе слепых и открытых клинических исследований более 18 000 пациентов прошли курс лечения секукинумабом по различным показаниям (бляшечный псориаз, псориатический артрит, аксиальный спондилоартрит и другие аутоиммунные заболевания), что является выборкой из 30 565 пациентов, получавших секукинумаб в течение нескольких лет. Из них более 11 700 пациентов получали секукинумаб в течение минимум одного года. Профиль безопасности секукинумаба одинаков независимо от показаний.
Таблица 3. Перечень нежелательных реакций в клинических исследованиях1 и постмаркетинговом опыте
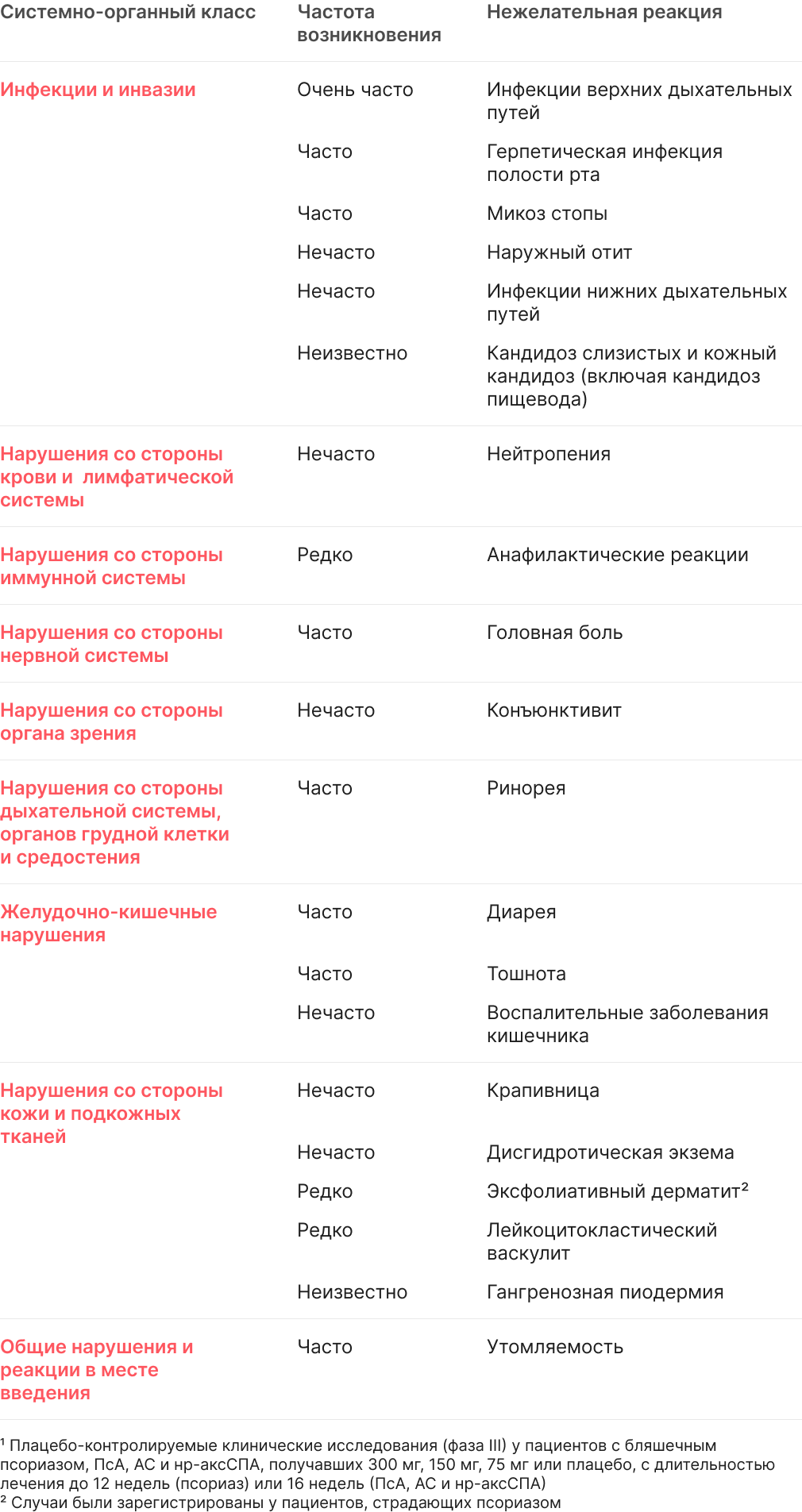
В плацебо-контролируемом периоде клинических исследований бляшечного псориаза (итого 1382 пациента, получавших секукинумаб, и 694 пациента, получавших плацебо, в течение 12 недель) сообщалось о выявлении инфекции у 28,7% пациентов, получавших секукинумаб, по сравнению с 18,9% пациентов, получавших плацебо. Большинство инфекций отнесены к несерьезным и легким или умеренным формам инфекций верхних дыхательных путей, например, назофарингит, которые не требовали прекращения лечения. Выявлен рост числа случаев кандидоза слизистых или кожного кандидоза, связанный с механизмом действия; выявленные случаи были отнесены к несерьезным, от легкой до умеренной степени тяжести, реагирующим на стандартное лечение и не требующим прекращения лечения. Серьезные инфекции зафиксированы у 0,14% пациентов, получавших секукинумаб, и у 0,3% пациентов, получавших плацебо (см раздел 4.4).
За весь период лечения (итого 3430 пациентов, получавших секукинумаб, вплоть до 52 недель для большинства пациентов) сообщалось о развитии инфекции у 47,5% пациентов, получавших секукинумаб (0,9 на пациентогод наблюдения). Серьезные инфекции были зарегистрированы у 1,2% пациентов, получавших секукинумаб (0,015 на пациенто-год наблюдения).
Частота инфекций, наблюдаемых в клинических исследованиях у пациентов с псориатическим артритом и аксиальным спондилоартритом (анкилозирующим спондилитом и нерентгенологическим аксиальным спондилоартритом), была идентична таковой в ходе исследований псориаза.
Нейтропения
В клинических исследованиях псориаза фазы III нейтропения чаще наблюдалась при использовании секукинумаба, чем при использовании плацебо, но в большинстве случаев была легкой, переходной и обратимой. Об уровне нейтропении <1,0-0,5x109 /л (3 класс в соответствии с Общими терминологическими критериями оценки нежелательных явлений (3 класс в соответствии с CTCAE)) сообщалось у 18 из 3430 (0,5%) пациентов, получавших секукинумаб, вне зависимости от дозы и без временных соотношений с инфекциями в 15 из 18 случаев.
Сообщения о случаях более тяжелых форм нейтропении отсутствуют. В оставшихся 3 случаях сообщалось о несерьезных инфекциях с обычным ответом на стандартную терапию, не требующих прекращения приема секукинумаба.
Частота возникновения нейтропении при псориатрическом артрите и аксиальном спондилоартрите (анкилозирующем спондилите и нерентгенологическом аксиальном спондилоартрите) была идентична таковой при псориазе.
Сообщалось о редких случаях нейтропении <0,5x109 /л (4 класс в соответствии с CTCAE)
Иммуногенность
В клинических исследованиях на пациентах с псориазом, псориатическим артритом и аксиальным спондилоартритом (анкилозирующим спондилитом и нерентгенологическим аксиальным спондилоартритом) менее чем у 1% пациентов, получавших секукинумаб, развились антитела к секукинумабу вплоть до 52 недель лечения. Около половины возникших после начала лечения антител на лекарственный препарат были нейтрализованы, но это не было связано с потерей эффективности или фармакокинетическими аномалиями.
Пациенты детского возраста
Нежелательные эффекты у пациентов детского возраста старше 6 лет с бляшечным псориазом
Безопасность секукинумаба оценивалась в двух исследованиях фазы III у детей с бляшечным псориазом. Первым исследованием (исследование 1 у пациентов детского возраста) было двойное слепое, плацебоконтролируемое исследование с участием 162 пациентов в возрасте от 6 до 18 лет с тяжелыми формами бляшечного псориаза. Вторым исследованием (исследование 2 у пациентов детского возраста) было открытое исследование с участием 84 пациентов в возрасте от 6 до 18 лет с умеренными или тяжелыми формами бляшечного псориаза.
Профиль безопасности, представленный в этих двух исследованиях, соответствовал профилю безопасности, описанному у взрослых пациентов с бляшечным псориазом.
Нежелательные эффекты у пациентов детского возраста с ЮИА
Безопасность секукинумаба также оценивали в исследовании фазы III у 86 пациентов с ювенильным идиопатическим артритом, а именно ЭАА и ЮПА, в возрасте от 2 до менее 18 лет. Профиль безопасности, представленный в этом исследовании, соответствовал профилю безопасности, описанному у взрослых пациентов.
Важно сообщать о подозреваемых нежелательных реакциях после регистрации ЛП с целью обеспечения непрерывного мониторинга соотношения «польза – риск» ЛП. Медицинским работникам рекомендуется сообщать о любых подозреваемых нежелательных реакциях ЛП через национальную систему сообщения о нежелательных реакциях РК.
РГП на ПХВ «Национальный центр экспертизы лекарственных средств и медицинских изделий» Комитета медицинского и фармацевтического контроля Министерства здравоохранения Республики Казахстан http://www.ndda.kz
Дозы до 30 мг/кг (приблизительно от 2000 до 3000 мг) вводились в клинических исследованиях внутривенно, без дозолимитирующей токсичности. В случае передозировки рекомендуется контролировать возникновение у пациента любых признаков или симптомов нежелательных реакций и немедленно назначить соответствующее симптоматическое лечение.
5. Фармакологические свойства
Фармакотерапевтическая группа: Иммуносупрессанты. Интерлейкина ингибиторы. Секукинумаб.
Код АТХ L04AC10
Секукинумаб представляет собой полностью человеческое моноклональное антитело IgG1/κ-класса, которое селективно связывается и нейтрализует провоспалительный цитокин интерлейкин17A (IL-17A). Секукинумаб обнаруживает IL-17A и ингибирует его взаимодействия с рецептором IL-17, который экспрессируется в различных типах клеток, включая кератиноциты. В результате секукинумаб ингибирует высвобождение провоспалительных цитокинов, хемокинов и медиаторов повреждения тканей и снижает опосредованное IL17A аутоиммунное участие в развитии воспалительных заболеваний.
Клинически значимые уровни секукинумаба проникают в кожу и уменьшают количество маркеров местного воспаления. Как следствие, лечение секукинумабом уменьшает покраснение, уплотнение и шелушение кожи, характерное для поражений бляшечным псориазом. IL-17A является естественным цитокином, который участвует в нормально протекающих реакциях воспаления и иммунного ответа. IL-17A играет ключевую роль в патогенезе бляшечного псориаза, псориатического артрита и аксиального спондилоартрита (анкилозирующего спондилита и нерентгенологического аксиального спондилоартрита) и активируется в поврежденной коже, в отличие от неповрежденной кожи, у пациентов с бляшечным псориазом и в синовиальной ткани у пациентов с псориатическим артритом.
Количество клеток, продуцирующих IL-17, было также существенно выше в субхондральных отделах костной ткани фасеточных суставов у пациентов с анкилозирующим спондилитом. Кроме этого, было обнаружено увеличение количества лимфоцитов, продуцирующих IL-17A, у пациентов с нерентгенологическим аксиальным спондилоартритом.
Была продемонстрирована эффективность ингибирования IL-17A, при лечении анкилозирующего спондилита, тем самым утверждая ключевую роль этого цитокина при аксиальном спондилоартрите.
Уровни общего IL-17A в сыворотке (свободного и связанного с секукинумабом IL-17A) первоначально увеличились у пациентов, получавших секукинумаб. За повышением последовало медленное снижение вследствие замедления клиренса IL-17A, связанного с секукинумабом, подтверждающее, что секукинумаб селективно связывается со свободным IL-17A, который играет ключевую роль в патогенезе бляшечного псориаза.
В исследовании у пациентов с бляшечным псориазом после одной-двух недель лечения секукинумабом значительно снижались инфильтрация эпидермиса нейтрофилами и количество различных ассоциированных с ними маркеров, которое часто повышено в пораженных участках кожи у данных пациентов.
Выявили, что секукинумаб уменьшал (в течение 1-2 недель после лечения) уровни С-реактивного белка, который являлся маркером воспаления.
Безопасность и эффективность секукинумаба были оценены в четырех рандомизированных, двойных слепых, плацебо-контролируемых исследованиях 3-ей фазы у пациентов с умеренным или тяжелым бляшечным псориазом, которым назначалась фототерапия или системная терапия [ERASURE, FIXTURE, FEATURE, JUNCTURE].
Эффективность и безопасность дозировок секукинумаба в 150 мг и 300 мг оценивались в сравнении с плацебо или этанерцептом. Кроме того, в одном исследовании длительный курс лечения оценивался в сравнении с «повторяющимся по мере необходимости» режимом лечения [SCULPTURE].
Из 2403 пациентов, которые были включены в плацебо-контролируемые исследования, 79% ранее не получали лечение биологическими препаратами, у 45% отмечалась неудача в лечении небиологическими препаратами, а у 8% отмечалась неудача терапии биологическими препаратами (6% с отсутствием эффекта при лечении анти-ФНО, 2% с отсутствием эффекта при использовании анти-р40). Примерно у 15- 25% пациентов в исследованиях 3-ей фазы отмечался псориатический артрит (ПА) в начале исследования.
В исследовании псориаза 1 (ERASURE) участвовали 738 пациентов.
Пациенты, рандомизированные в группу лечения секукинумабом, получали дозу в 150 мг или 300 мг в 0, 1, 2, 3 и 4 неделю, а затем ту же дозу каждый месяц. Исследование псориаза 2 (FIXTURE) оценивало 1306 пациентов.
Пациенты, рандомизированные в группу лечения секукинумабом, получали дозу в 150 мг или 300 мг в 0, 1, 2, 3 и 4 неделю, а затем ту же дозу каждый месяц. Пациенты, рандомизированные в группу лечения этанерцепта, получали дозу в 50 мг два раза в неделю в течение 12 недель с последующей дозой в 50 мг каждую неделю. В обоих исследованиях (1 и 2) пациенты, рандомизированные для получения плацебо, у которых отсутствовал ответ на 12-й неделе, были переведены на секукинумаб (150 мг или 300 мг) на 12, 13, 14, и 15 неделе, а затем ту же дозу каждый месяц, начиная с 16 недели. Наблюдение пациентов проводилось до 52 недель после первого введения исследуемого препарата.
В исследовании псориаза 3 (FEATURE) оценивали 177 пациентов с использованием предварительно заполненных шприцов в сравнении с плацебо после 12 недель лечения для оценки безопасности, переносимости и удобства самостоятельного введения секукинумаба с помощью предварительно заполненного шприца. Исследование псориаза 4 (JUNCTURE) оценивало 182 пациента с использованием предварительно заполненных шприц-ручек по сравнению с плацебо после 12 недель лечения для оценки безопасности, переносимости и удобства применения секукинумаба с помощью предварительно заполненной шприц-ручки. В обоих исследованиях (3 и 4) пациенты, рандомизированные для получения секукинумаба, получали дозу в 150 мг или 300 мг в 0, 1, 2, 3 и 4 неделю, а затем в той же дозе каждый месяц.
Пациенты были также рандомизированы для получения плацебо в 0, 1, 2, 3 и 4 неделю, а затем в той же дозе каждый месяц.
Исследование 5 (SCULPTURE) оценивало 966 пациентов. Все пациенты, получавшие секукинумаб в дозе 150 мг или 300 мг в 0, 1, 2, 3, 4, 8 и 12 неделю, а затем были рандомизированы для получения либо поддерживающего режима в той же дозе каждый месяц, начиная с 12 недели, либо «повторения лечения при необходимости» в той же дозе. У пациентов, рандомизированных для получения «повторного лечения при необходимости», не удалось достичь надлежащего ответа, следовательно, рекомендуется длительный режим с ежемесячным введением.
Комбинированные основные конечные точки в исследованиях плацебо и активных контролируемых исследованиях отображали долю пациентов, достигших индекса площади и тяжести псориаза PASI 75, и «чистого» или «почти чистого» ответа по сравнению с плацебо по системе глобальной оценки исследователя 2011 на 12 неделе (см. таблицу 4 и 5). Доза в 300 мг обеспечила лучшее очищение кожи, особенно для «чистой» или «почти чистой» кожи, наряду с основными конечными точками индекса площади и тяжести псориаза PASI 90 и 100 и ответом 0 и 1 по системе глобальной оценки исследователя (IGA) 2011, во всех исследованиях с наблюдаемым пиковым эффектом на 16 неделе, следовательно, именно эта доза является рекомендуемой.
Таблица 4. Обзор индексов площади и тяжести поражения псориазом PASI 50/75/90/100 и «чистого» или «почти чистого» клинического ответа по системе глобальной оценки исследователя* 2011 в исследованиях псориаза 1, 3 и 4 (ERASURE, FEATURE и JUNCTURE)

Таблица 5. Обзор клинических ответов в исследовании псориаза 2 (FIXTURE)

В дополнительном исследовании на пациентах с псориазом (CLEAR) оценили состояние 676 пациентов. Секукинумаб в дозе 300 мг соответствовал первичным и вторичным конечным точкам, демонстрируя превосходство над устекинумабом с учетом ответа PASI 90 на неделе 16 (первичная конечная точка), скорости возникновения ответа PASI 75 на неделе 4 и долгосрочного ответа PASI 90 на неделе 52. Повышенную эффективность секукинумаба в сравнении с устекинумабом для конечных точек PASI 75/90/100 и баллы 0 или 1 по Системе глобальной оценки 2011 г. («чистый» или «почти чистый») наблюдали на начальном этапе и далее до недели 52.
Таблица 6. Обзор клинических ответов в исследовании CLEAR

Секукинумаб демонстрировал эффективность при лечении пациентов, ранее не получавших системное лечение, бионаивных, получавших лечение анти-ФНО и биологическими препаратами, и пациентов, у которых лечение анти-ФНО и биологическими препаратами было неэффективным. Улучшения индекса PASI 75 у пациентов с сопутствующим псориатическим артритом на исходном уровне было сходно с результатами для пациентов из общей популяции бляшечного псориаза.
Секукинумаб ассоциировался с быстрым достижением эффективности с сокращением на 50% среднего значения индексa площади и тяжести поражения псориазом PASI через 3 недели для дозы 300 мг
Рис. 1. Динамика процентных изменений от исходного уровня среднего значения индекса площади и тяжести поражения псориазом PASI в исследовании 1 (ERASURE)  Специфические локализации/формы бляшечного псориаза
Специфические локализации/формы бляшечного псориаза
В двух дополнительных плацебо-контролируемых исследованиях отмечалось улучшение состояния ногтей при псориазе (TRANSFIGURE, 198 пациентов) и улучшение состояния при ладонно-подошвенном бляшечном псориазе (GESTURE, 205 пациентов). В исследовании TRANSFIGURE секукинумаб обладал большей эффективностью по сравнению с плацебо на неделе 16 (46,1% для 300 мг, 38,4% для 150 мг и 11,7% для плацебо) с учетом значительного улучшения по сравнению с исходным уровнем Индекса тяжести псориаза ногтей (NAPSI %) у пациентов с умеренной и тяжелой формой бляшечного псориаза с поражением ногтевой пластины. В исследовании GESTURE секукинумаб обладал большей эффективностью по сравнению с плацебо на неделе 16 (33,3% для 300 мг, 22,1% для 150 мг и 1,5% для плацебо) с учетом значительного улучшения ответа в виде 0 или 1 балла по системе глобальной оценки исследователя (ppIGA) («чистый» или «почти чистый») у пациентов с умеренной и тяжелой формой ладонно-подошвенного бляшечного псориаза.
В плацебо-контролируемом исследовании оценили состояние 102 пациентов с умеренной и тяжелой формой псориаза волосистой части головы, определяемого как значение индекса тяжести поражения псориазом волосистой части головы (PSSI) ≥12, баллы 3 или выше по системе глобальной оценки исследователя только для волосистой части головы IGA 2011 и поражение площади поверхности кожи волосистой части головы не менее 30%. Секукинумаб 300 мг обладал большей эффективностью по сравнению с плацебо на неделе 12 с учетом значительного улучшения значения PSSI не менее чем на 90% (52,9% vs 2,0%) и значения 0 или 1 по системе глобальной оценки исследователя 2011 г. только для волосистой части головы (56,9% vs 5,9%). Улучшение для обеих конечных точек сохранялось у пациентов из группы приема секукинумаба, которые продолжили лечение до недели 24.
Качество жизни/оценка результатов пациентами
Статистически значимые результаты на 12 неделе (исследования 1-4) от исходного уровня, в сравнении с плацебо, демонстрировались в соответствии с дерматологическим индексом качества жизни (DLQI).
Средние значения снижения (улучшения) DLQI от исходного уровня варьировались от -10,4 до -11,6 для секукинумаба 300 мг, от -7,7 до -10,1 для секукинумаба 150 мг, в сравнении с диапазоном от -1,1 до -1,9 для плацебо на 12 неделе. Эти улучшения сохранялись до 52 недель (Исследования 1 и 2).
Сорок процентов участников исследования 1 и 2 заполняли дневник симптомов псориаза (Psoriasis Symptom Diary©). Участники в каждом из этих исследований, заполнившие дневник, подтвердили статистически значимые улучшения на 12 неделе от исходного уровня в сравнении с сообщаемыми ими симптомами зуда, боли и шелушения.
Статистически значимые улучшения на неделе 4 относительно исходных значений у пациентов, получавших лечение секукинумабом, по сравнению с пациентами, получавшими устекинумаб (CLEAR), были продемонстрированы на примере значений индекса качества жизни при заболеваниях кожи и сохранялись до недели 52.
Статистически значимые улучшения сообщаемых пациентами симптомов и таких симптомов, как зуд, боль и образование псориатических чешуек на неделе 16 и неделе 52 (CLEAR) были занесены в дневник симптомов псориаза у пациентов, получавших секукинумаб, по сравнению с пациентами, получавшими устекинумаб.
В исследовании псориаза волосистой части головы были показаны статистически значимые улучшения (уменьшение) на неделе 12 от исходного уровня сообщаемых пациентами таких признаков и симптомов, как зуд, боль и образование псориатических чешуек по сравнению с плацебо.
Диапазон дозировки при бляшечном псориаз
В рандомизированном двойном слепом многоцентровом исследовании оценивали две схемы введения поддерживающей дозы (300 мг каждые 2 недели и 300 мг каждые 4 недели) с помощью предварительно заполненного шприца по 150 мг у 331 пациента с массой тела ≥ 90 кг с псориазом средней и тяжелой степени. Пациенты были рандомизированы 1:1 следующим образом:
- секукинумаб 300 мг на 0, 1, 2, 3 и 4 неделях, а затем в той же дозе каждые 2 недели до 52-й недели (n = 165).
- секукинумаб 300 мг на 0, 1, 2, 3 и 4 неделях, а затем в той же дозе каждые 4 недели до 16-й недели (n = 166).
- Пациенты, рандомизированные для получения секукинумаба в дозе 300 мг каждые 4 недели, у которых на 16-й неделе был ответ PASI 90, продолжали получать тот же режим дозирования до 52-й недели. Пациенты, рандомизированные для получения секукинумаба в дозе 300 мг каждые 4 недели, у которых на 16-й неделе не было ответа по критериям PASI 90, либо продолжали получать тот же режим дозирования, либо были переведены на получение секукинумаба в дозе 300 мг каждые 2 недели до 52-й недели.
В целом показатели частоты ответа эффективности в группе, получавшей лечение по схеме каждые 2 недели, были выше по сравнению с группой, получавшей лечение по схеме каждые 4 недели (таблица 7).
Таблица 7. Обзор клинических ответов в исследовании диапазона дозировки при бляшечном псориазе*

У пациентов, не достигших ответа PASI 90 на 16 неделе, которым была повышена доза секукинумаба до 300 мг каждые 2 недели, наблюдалось улучшение показателей частоты ответа PASI 90 по сравнению с теми, кто продолжал получать секукинумаб в дозе 300 мг каждые 4 недели, в то время как показатели частоты ответа 0/1 по IGA mod 2011 оставались стабильными с течением времени в обеих группах лечения.
Профили безопасности двух режимов дозирования препарата Козэнтикс 300 мг, вводимого каждые 4 недели, и Козэнтикс 300 мг, вводимого каждые 2 недели, у пациентов с массой тела ≥ 90 кг были сопоставимы и соответствовали профилю безопасности, о котором сообщалось у пациентов с псориазом.
Безопасность и эффективность секукинумаба оценили в трех рандомизированных, двойных слепых, плацебо-контролируемых исследованиях III фазы у 1999 пациентов с активным псориатическим артритом (≥3 отекших и ≥3 болезненных суставов), несмотря на прием нестероидных противовоспалительных средств (НПВС), кортикостероидов или болезнь-модифицирующих антиревматических препаратов. В эти исследования были включены пациенты с каждым подтипом ПсА, включая полиартрит без признаков ревматоидных узлов, спондилит с периферическим артритом, ассиметрический периферический артрит, поражение дистальных межфаланговых суставов и мутилирующий артрит. Пациенты, участвовавшие в этих исследованиях, страдали ПсА минимум в течение 5 лет. Большинство пациентов имели активные псориатические повреждения кожи или документально подтвержденный псориаз в анамнезе. Свыше 61% и 42% пациентов с ПсА имели энтезит и дактилит на исходном уровне, соответственно. Во всех исследованиях первичной конечной точкой явилось значение ACR 20 (Американская коллегия ревматологии).
Для исследования псориатического артрита 1 (исследование ПсA 1) и исследования псориатического артрита 2 (исследование ПсA 2) первичная конечная точка была на 24 неделе. Для исследования псориатического артрита 3 (исследование ПсA 3) первичная конечная точка была на 16 неделе, а ключевая вторичная конечная точка, измененная в сравнении с исходными значениями согласно модифицированной общей системе баллов по Sharp (mTSS), на 24 неделе.
В исследованиях ПсА 1, ПсА 2 и ПсА 3 29%, 35% и 30% пациентов соответственно ранее получали лечение ингибиторами ФНО-альфа и прекратили принимать ингибитор ФНО-альфа в связи с отсутствием эффективности или непереносимостью (пациенты, получающие ингибиторы ФНО-альфа).
В исследовании ПсА 1 (FUTURE 1) оценили состояние 606 пациентов, 60,7% из которых одновременно принимали метотрексат. Пациенты, рандомизированные в группу лечения секукинумабом, получали 10 мг/кг в/в в 0, 2 и 4 неделю, а затем 75 мг или 150 мг п/к каждый месяц, начиная с 8 недели. Пациенты, рандомизированные для получения плацебо, у которых отсутствовал ответ на 16-й неделе (ранняя терапия), и другие пациенты из группы плацебо на неделе 24 были переведены на секукинумаб (75 мг или 150 мг п/к), а затем ту же дозу каждый месяц.
В исследовании ПсА 2 (FUTURE 2) оценили состояние 397 пациентов, 46,6% из которых одновременно принимали метотрексат. Пациенты, рандомизированные в группу лечения секукинумабом, получали дозу в 75 мг, 150 мг или 300 мг п/к в 0, 1, 2, 3 и 4 неделю, а затем ту же дозу каждый месяц. Пациенты, рандомизированные для получения плацебо, у которых отсутствовал ответ на 16-й неделе, были переведены на секукинумаб (150 мг или 300 мг подкожно) на 16 неделе (ранняя терапия), а затем получали ту же дозу каждый месяц.
Пациенты, рандомизированные для получения плацебо, у которых присутствовал ответ на 16-й неделе, были переведены на секукинумаб (150 мг или 300 мг подкожно) на неделе 24, а затем получали ту же дозу каждый месяц.
В исследовании ПсА 3 (FUTURE 5) оценили состояние 996 пациентов, 50,1% из которых одновременно принимали метотрексат. Пациенты, рандомизированные в группу лечения секукинумабом, получали дозу 150 мг, 300 мг или плацебо подкожно в 0, 1, 2, 3 и 4 неделю, а затем такую же дозу каждый месяц или раз в месяц инъекцию секукинумаба 150 мг (без нагрузки). Пациенты, рандомизированные для получения плацебо, у которых отсутствовал ответ на 16 неделе (ранняя терапия), были переведены на секукинумаб (150 мг или 300 мг подкожно) на 16 неделе, а затем получали ту же дозу каждый месяц. Пациенты, рандомизированные для получения плацебо, у которых был ответ на 16 неделе, были переведены на секукинумаб (150 мг или 300 мг подкожно) на неделе 24, а затем получали ту же дозу каждый месяц.
Признаки и симптомы
Лечение секукинумабом позволило значительно улучшить показатели активности заболевания по сравнению с плацебо на 16 и 24 неделях (см. таблицу 8).
Таблица 8. Клинический ответ в исследованиях ПсА 2 и ПсА 3 на 16 и 24 неделях

Начало действия секукинумаба проявляется на неделе 2. Статистически значимое различие в ACR 20 в сравнении с плацебо было достигнуто на неделе 3.
Процент пациентов, достигших ответа со значением ACR 20 с учетом визита, показан на рис. 2.
Рис. 2. Клинический ответ ACR20 в исследовании ПсА 2 до 52 недели 
Похожие ответы для первичных и ключевых вторичных конечных точек были обнаружены у пациентов с ПсА, независимо от того, получали ли они одновременно лечение метотрексатом или нет. В исследовании ПсА 2 у пациентов, получавших одновременно секукинумаб и метотрексат, отмечался повышенный ответ ACR 20 (47,7% и 54,4% для 150 мг и 300 мг, соответственно, в сравнении с плацебо 20,0%) и ответ ACR 50 (31,8% и 38,6% для 150 мг и 300 мг, соответственно, в сравнении с 8,0% для плацебо). У пациентов, получавших секукинумаб без одновременного применения метотрексата, отмечался повышенный ответ ACR 20 (53,6% и 53,6% для 150 мг и 300 мг, соответственно, в сравнении с 10,4% для плацебо) и ответ ACR 50 (37,5% и 32,1% для 150 мг и 300 мг, соответственно, в сравнении с 6,3% для плацебо).
В исследовании ПсА 2 у пациентов, которые ранее не получали антиФНО-альфа терапию и у пациентов с неадекватным ответом на антиФНО-альфа терапию отмечался значительно более повышенный ответ ACR 20 в сравнении с плацебо на неделе 24 и незначительно более высокий ответ в группе пациентов, которые ранее не получали антиФНО-альфа терапию (без анти-ФНО-альфа терапии: 64% и 58% для 150 мг и 300 мг, соответственно, в сравнении с плацебо 15,9%; у пациентов с неадекватным ответом на анти-ФНО-альфа терапию: 30% и 46% для 150 мг и 300 мг, соответственно, в сравнении с 14,3% для плацебо). В подгруппе пациентов с неадекватным ответом на анти-ФНО-альфа терапию при введении препарата только в дозе 300 мг обнаружили значительно более повышенную частоту ACR 20 по сравнению с плацебо (p <0,05) и клинически значимую выгоду при введении препарата в дозе выше 150 мг в множественные вторичные ключевые точки. Улучшение индекса PASI 75 отмечалось в обеих подгруппах, а в подгруппе 300 мг выявили статистически значимое улучшение у пациентов с неадекватным ответом на анти-ФНО-альфа терапию.
Количество пациентов с поражением осевого скелета при ПсА было слишком маленьким, чтобы можно было провести значимую оценку. Были продемонстрированы улучшения по всем компонентам значений ACR, включая оценку боли пациентом. В исследовании ПсА 2 процент пациентов, достигших модифицированного ответа с учетом критериев ответа при ПсА, был выше в группе приема секукинумаба (59,0% и 61,0% для 150 мг и 300 мг, соответственно) в сравнении с плацебо (26,5%) на неделе 24. В исследовании ПсА 1 и ПсА 2 эффективность сохранялась до недели 104.
В исследовании ПсА 2 из 200 пациентов, изначально рандомизированных в группу приема секукинумаба в дозе 150 мг и 300 мг, 178 (89%) пациентов по-прежнему получали лечение на неделе 52. Из 100 пациентов, рандомизированных для приема секукинумаба в дозе 150 мг, у 64, 39 и 20 пациентов отмечался ответ со значением ACR 20/50/70, соответственно. Из 100 пациентов, рандомизированных для приема секукинумаба в дозе 300 мг, у 64, 44 и 24 пациентов отмечался ответ со значением ACR 20/50/70, соответственно.
Рентгенографический ответ
В исследовании ПсA 3 ингибирование прогрессирования разрушения структуры оценили рентгенографически и выразили как изменение в модифицированном общем счёте Шарпа (mTSS) и его компонентов, изменение баллов по шкале оценки эрозии (ES) и сужения суставной щели (JSN). Рентгенограммы кистей, запястий и стоп были выполнены в начале исследования, на 16 и/или 24 неделях и оценивались независимо, по крайней мере, двумя специалистами, которые не знали ни группу пациентов, ни количество посещений. Лечение секукинумабом в дозах 150 и 300 мг значительно ингибировало скорость прогрессирования повреждения периферических суставов по сравнению с лечением плацебо, которое измерялось в сравнении с исходными показателями mTSS на 24 неделе (таблица 9).
Ингибирование прогрессирования разрушения структуры также оценивали в исследовании ПсА 1 на 24 и 52 неделях в сравнении с исходными показателями. Данные, полученные на неделе 24, представлены в таблице 8.
Таблица 9. Изменение в модифицированном общем счете Шарпа у пациентов с псориазом

В исследовании ПсА 1 ингибирование повреждения структуры сохранялось при лечении секукинумабом до 52 недель.
В исследовании PsA 3 процент пациентов без прогрессирования заболевания (определяемых как изменение от базового уровня mTSS <0,5) от рандомизации до 24 недели составил 80,3%, 88,5% и 73,6% для секукинумаба 150 мг, 300 мг и плацебо, соответственно. Эффект ингибирования структурного повреждения наблюдался у пациентов, не получавших анти-ФНОα, у пациентов с неадекватным ответом на анти-ФНОα, и у пациентов, получавших одновременно метотрексат и без него.
В исследовании ПсА 1 процент пациентов без прогрессирования заболевания (определяемое как изменение с исходного уровня значения mTSS на ≤0,5) с рандомизации до недели 24 составил 82,3% в группе пациентов, получающих секукинумаб внутривенно в дозе 10 мг/кг или подкожно в дозе 150 мг и 75,7% в группе, получающих плацебо. Процент пациентов без прогрессирования заболевания с недели 24 до недели 52 в группе пациентов, получающих секукинумаб внутривенно в дозе 10 мг/кг, а затем - подкожно в дозе 75 мг или 150 мг, а также у пациентов в группе плацебо, которые получали препарат в дозе 150 мг подкожно каждые 4 недели на неделе 16 или неделе 24 составил 85,7 и 86,8%, соответственно.
Поражение осевого скелета при ПсА
В рандомизированном двойном слепом плацебо-контролируемом исследовании (MAXIMIZE) оценивалась эффективность секукинумаба у 485 пациентов с ПсА с поражением осевого скелета.
Эти пациенты не получали биологические препараты и неадекватно отвечали на терапию НПВП. Основной показатель - улучшение как минимум на 20% критериев шкалы Международного общества по изучению спондилоартрита (ASAS 20) на 12 неделе терапии. Лечение секукинумабом 300 мг и 150 мг при сравнении с лечением плацебо также привело к более выраженной положительной динамике клинических проявлений поражения осевого скелета (включая уменьшение боли в спине по сравнению с исходным уровнем) и к улучшению функционального статуса (см. Таблицу 10).
Таблица 10. Клинический ответ в исследовании MAXIMISE на 12-й неделе

Улучшение ASAS 20 и ASAS 40 для обеих доз секукинумаба наблюдалось к 4 неделе и сохранялось до 52 недель
Функциональный статус и качество жизни, обусловленное состоянием здоровья
В исследовании ПсА 2 и ПсА 3 у пациентов, получавших секукинумаб в дозе 150 мг (p=0,0555 и p <0.0001) и 300 мг (p=0,0040 и p <0.0001), выявлено улучшение функционального статуса по сравнению с пациентами, получавшими плацебо в соответствии с опросником для оценки состояния здоровья — индекс инвалидизации (HAQ-DI) на неделе 24 и 16. Улучшение баллов HAQ-DI наблюдалось независимо от предыдущего воздействия анти-ФНО-препаратов. Похожие ответы наблюдались в исследовании ПсА 1.
Пациенты, получавшие секукинумаб, сообщали о значительных улучшениях качества жизни, связанного с состоянием здоровья в соответствии с баллами опросника SF-36 для оценки индекса физического здоровья (SF-36 PCS) (p<0,001). Также обнаружили статистически значимые улучшения эксплоративных конечных точек по данным Опросника функциональной оценки терапии хронического заболевания — усталость (FACIT-F) для групп 150 мг и 300 мг по сравнению с плацебо (7,97, 5,97 в сравнении с 1,63, соответственно) и эти улучшения сохранялись до 104 недели в исследовании ПсA 2.
Похожие ответы наблюдались в исследовании ПсА 1, а эффективность сохранялась до недели 52.
Безопасность и эффективность секукинумаба оценили в трех рандомизированных, двойных слепых, плацебо-контролируемых исследованиях III фазы у 816 пациентов с активным анкилозирующим спондилитом (АС) с Батским индексом активности заболевания при анкилозирующем спондилите (BASDAI) ≥4, несмотря на прием нестероидных противовоспалительных средств, кортикостероидов или болезнь-модифицирующих антиревматических препаратов. Пациентам в исследованиях АС 1 и АС 2 диагностировали АС, в среднем, в течение 2,7-5,8 лет. В обоих исследованиях первичная конечная точка составила не менее 20% улучшения критериев по Шкале оценки Международного общества по изучению спондилоартритов (ASAS 20) на неделе 16.
В исследовании анкилозирующего спондилита 1 (исследование АС 1), исследовании анкилозирующего спондилита 2 (исследование АС 2) и исследовании анкилозирующего спондилита 3 (исследование АС 3) 27,0%, 38,8% и 23,5% пациентов соответственно ранее получали лечение ингибиторами ФНО-альфа и прекратили принимать ингибитор ФНО-альфа в связи с отсутствием эффективности или непереносимостью (пациенты с неадекватным ответом на терапию ингибиторами ФНО-альфа).
В исследовании АС 1 (MEASURE 1) оценили состояние 371 пациента, 14,8% и 33,4% из которых одновременно принимали метотрексат или сульфасалазин, соответственно. Пациенты, рандомизированные в группу лечения секукинумабом, внутривенно получали 10 мг/кг в/в в 0, 2 и 4 неделю, а затем — подкожно 75 мг или 150 мг каждый месяц, начиная с недели 8. Пациенты, рандомизированные для получения плацебо, у которых отсутствовал ответ на 16-й неделе (ранняя терапия), и другие пациенты из группы плацебо на неделе 24 были переведены на секукинумаб (75 мг или 150 мг п/к), а затем получали ту же дозу каждый месяц.
В исследовании АС 2 (MEASURE 2) оценили состояние 219 пациента, 11,9% и 14,2% из которых одновременно принимали метотрексат или сульфасалазин, соответственно. Пациенты, рандомизированные в группу лечения секукинумабом, получали дозу в 75 мг или 150 мг п/к в 0, 1, 2, 3 и 4 неделю, а затем ту же дозу каждый месяц. Пациенты, рандомизированные для получения плацебо на начальном этапе, прошли повторную рандомизацию на 16-й неделе для получения секукинумаба (75 мг или 150 мг п/к) каждый месяц.
В исследовании 3 (MEASURE 3) оценивали 226 пациентов, из которых 13,3% и 23,5% использовали сопутствующий метотрексат или сульфасалазин соответственно. Пациенты, рандомизированные в группу лечения секукинумабом, получали дозу 10 мг / кг внутривенно в 0, 2 и 4 недели, а затем подкожно 150 мг или 300 мг каждый месяц.
На 16 неделе пациенты, которые были рандомизированы в группу плацебо в начале исследования, были повторно рандомизированы для получения секукинумаба (150 мг или 300 мг подкожно) каждый месяц. Первичной конечной точкой был ASAS 20 на 16 неделе.
Пациенты не знали о режиме лечения до 52 недели, и исследование продолжалось до 156 недели.
Признаки и симптомы
В исследовании АС 2 лечение секукинумабом в дозе 150 мг привело к более значительному улучшению показателей активности заболевания по сравнению с плацебо на неделе 16 (см. таблицу 11).
Таблица 11. Клинический ответ в исследовании АС 2 на неделе 16

Начало действия секукинумаба в дозе 150 мг проявляется на неделе 1 для ASAS 20 и неделе 2 для ASAS 40 (эффективнее плацебо) в исследовании АС 2.
Ответы для ASAS 20 улучшились на неделе 16 как у пациентов, не получавших анти-ФНО-альфа препараты (68,2% в сравнении с 31,1%, p <0,05), так и у пациентов с неадекватным ответом на терапию анти-ФНО-альфа препаратами (50,0% в сравнении с 24,1%; p <0,05) в группе секукинумаба 150 мг по сравнению с плацебо, соответственно. В исследованиях АС 1 и АС 2 у пациентов, получавших секукинумаб (150 мг в исследовании АС 2 и оба режима терапии в исследовании АС 1), показано значительное улучшение признаков и симптомов на неделе 16 с сопоставимой интенсивностью ответа и эффективностью, сохранявшихся до недели 52 как у пациентов, не получавших анти-ФНО-альфа препараты, так и у пациентов с неадекватным ответом на терапию анти-ФНОальфа препаратами. В исследовании АС 2 из 72 пациентов, изначально рандомизированных в группу приема секукинумаба в дозе 150 мг, 61 (84,7%) пациент по-прежнему получали лечение на неделе 52. Из 72 пациентов, рандомизированных для приема секукинумаба в дозе 150 мг, у 45 и 35 пациентов отмечался ответ ASAS 20/40, соответственно.
В исследовании АС 3 пациенты, получавшие секукинумаб (150 мг и 300 мг), продемонстрировали улучшение признаков и симптомов и имели сопоставимые ответы по эффективности независимо от дозы, которая превосходила плацебо на 16 неделе для первичной конечной точки (ASAS 20). В целом показатели эффективности ответа для группы 300 мг были последовательно выше по сравнению с группой 150 мг для вторичных конечных точек. Во время слепого периода ответы ASAS 20 и ASAS 40 составляли 69,7% и 47,6% для 150 мг и 74,3% и 57,4% для 300 мг на 52 неделе соответственно. Ответы ASAS 20 и ASAS 40 сохранялись до 156 недели (69,5% и 47,6% для 150 мг в сравнении с 74,8% и 55,6% для 300 мг). Более высокие показатели ответа в пользу 300 мг также наблюдались для ответа с частичной ремиссией ASAS (ASAS PR) на 16 неделе и сохранялись вплоть до недели 156. Большие различия в коэффициентах ответа, поддерживающие 300 мг над 150 мг, наблюдались у пациентов с неадекватным ответом на анти-ФНО-альфа препараты (n=36) по сравнению с пациентами, не получавшими анти-ФНО-альфа препараты (n = 114)
Подвижность позвоночника
У пациентов, получавших секукинумаб в дозе 150 мг, обнаружили улучшения подвижности позвоночника с учетом изменений баллов по шкале BASMI с исходного уровня на неделе 16 как в исследовании АС 1 (-0,40 в сравнении с -0,12 для плацебо; p=0,0114), так и в исследовании АС 2 (-0,51 в сравнении с -0,22 для плацебо; p=0,0533). Эти улучшения поддерживались до недели 52.
Функциональный статус и качество жизни, обусловленное состоянием здоровья
В исследовании АС 1 и 2 пациенты, получавшие секукинумаб в дозе 150 мг, продемонстрировали улучшения в качестве жизни, связанном со здоровьем, согласно Опроснику по качеству жизни при АС (ASQoL) (p=0,001) и Опроснику SF-36 для оценки индекса физического здоровья (SF-36 PCS) (p <0,001). У пациентов, получавших секукинумаб в дозе 150 мг, также были показаны статистически значимые улучшения эксплоративных конечных точек функционального статуса в соответствии с Батским индексом функциональных нарушений АС (BASFI) в сравнении с плацебо (-2,15 в сравнении с -0,68), а также усталости в соответствии с Опросником функциональной оценки терапии хронического заболевания - усталость (FACIT-F) по сравнению с плацебо (8,10 в сравнении с 3,30). Эти улучшения поддерживались до недели 52.
Нерентгенологический аксиальный спондилоартрит (нр-аксСПА)
Безопасность и эффективность секукинумаба были оценены у 555 пациентов в одном рандомизированном двойном слепом плацебо-контролируемом исследовании фазы III (PREVENT), состоящем из двухлетней основной фазы и двухлетней фазы дополнительного лечения, у пациентов с активным нерентгенологическим аксиальным спондилоартритом (нр-аксСПА), удовлетворяющим критериям классификации Международного общества оценки спондилоартрита (ASAS) для аксиального спондилоартрита (аксСПА) без рентгенографического подтверждения изменений в крестцово-подвздошных суставах, которые соответствовали бы модифицированным Нью-Йоркскими критериями для анкилозирующего спондилита (АС). Включенные в исследование пациенты имели активное заболевание, определяемое как Батский индекс активности заболевания при анкилозирующем спондилоартрите (BASDAI) > 4, Визуальноаналоговая шкала (VAS) для общей боли в спине ≥ 40 (по шкале 0–100 мм), несмотря на текущую или предшествующую терапию нестероидными противовоспалительными средствами (НПВС) и повышение уровня С-реактивного белка (СРБ) и (или) признаки сакроилиита на магнитнорезонансной томографии (МРТ). Пациентам с данном исследовании был поставлен диагноз аксСПА в среднем в течение 2,1–3,0 года, 54% участников исследования были женского пола.
В исследовании PREVENT 9,7% пациентов ранее получали лечение ингибиторами ФНО-альфа и прекратили принимать ингибитор ФНО-альфа в связи с отсутствием эффективности или непереносимостью (пациенты с неадекватным ответом на терапию ингибиторами ФНО-альфа).
В исследовании PREVENT 9,9% и 14,8% пациентов одновременно принимали метотрексат или сульфасалазин соответственно. В двойной слепой период пациенты получали либо плацебо, либо секукинумаб, в течение 52 недель. Пациенты, рандомизированные в группу лечения секукинумабом, получали дозу в 150 мг п/к в 0, 1, 2, 3 и 4 неделю, а затем ту же дозу каждый месяц, или ежемесячные инъекции секукинумаба в дозировке 150 мг. Первичной конечной точкой было улучшение по крайней мере на 40% критериев по шкале оценки Международного общества по изучению спондилоартритов (ASAS 40) на неделе 16.
Симптомы и проявления:
В исследовании PREVENT лечение препаратом секукинумаб в дозировке 150 мг позволили значительно улучшить показатели активности заболевания по сравнению с плацебо на неделе 16. Эти измерения включают ASAS 40, ASAS 5/6, оценку BASDAI, BASDAI 50, высокочувствительный CRP (hsCRP), ASAS 20 и ответ частичной ремиссии ASAS по сравнению с плацебо (таблица 12). Ответы сохранялись до 52 недель.
Таблица 12. Клинический ответ в исследовании PREVENT на неделе 16

Начало действия секукинумаба в дозе 150 мг проявляется на неделе 3 для ASAS 40 у пациентов, ранее не получавших лечение анти-ФНО-альфапрепаратами (эффективнее плацебо) в исследовании PREVENT. Процент пациентов, достигших ответа со значением ASAS 40 с у пациентов, ранее не получавших лечение анти-ФНО-альфа учетом визита, показан на рис. 3.
Рис. 3. Клинический ответ ASAS 40 у пациентов, ранее не получавших лечение анти-ФНО-альфа, в исследовании PREVENT до недели 16 
Ответы для ASAS 40 улучшились на неделе 16 у пациентов с неадекватным ответом на терапию анти-ФНО-альфа в группе приема секукинумаба 150 мг по сравнению с плацебо соответственно.
Функциональный статус и качество жизни, обусловленное состоянием здоровья:
Пациенты, получавшие лечение секукинумабом в дозировке 150 мг, продемонстрировали статистически значимые улучшения к 16 неделе по сравнению с пациентами, получавшими плацебо, в отношении физической функции по оценке BASFI (неделя 16: –1,75 в сравнении с –1,01, p <0,05). Пациенты, получавшие лечение секукинумабом, продемонстрировали статистически значимые улучшения к 16 неделе по сравнению с пациентами, получавшими плацебо, в отношении качества жизни, связанного со здоровьем, измеренного по ASQoL (среднее изменение LS: неделя 16: 3,45 по сравнению с –1,84 p < 0,05) и индексу физического здоровья SF-36 (SF-36 PCS) (LS среднее изменение: неделя 16: 5,71 в сравнении с 2,93 p < 0,05). Эти улучшения поддерживались до недели 52.
Подвижность позвоночника:
Подвижность позвоночника оценивалась по BASMI вплоть до 16 недели. Численно большее улучшение было продемонстрировано у пациентов, получавших секукинумаб, по сравнению с пациентами, получавшими плацебо, на 4, 8, 12 и 16 неделе.
Ингибирование воспалительного процесса на магнитно-резонансной терапии (МРТ):
Признаки воспаления оценивали с помощью МРТ в начале и на 16 неделе и выражали в виде изменения по сравнению с исходным уровнем по Берлинской шкале отека крестцово-подвздошного сустава для крестцово-подвздошных суставов, а также по шкале ASspiMRI-a и Берлинской шкале оценки состояния позвоночника для позвоночника.
У пациентов, получавших лечение секукинумабом, наблюдалось ингибирование симптомов воспаления и крестцово-подвздошных суставов, и позвоночника. Среднее изменение по сравнению с исходным уровнем по Берлинской шкале отека крестцово-подвздошного сустава составило –1,68 для пациентов, получавших лечение секукинумабом дозировкой 150 мг (n = 180) по сравнению с –0,39 для пациентов, получавших плацебо (n = 174) (p < 0,05).
Было показано, что секукинумаб улучшает признаки и симптомы, а также качество жизни, связанное со здоровьем, у пациентов детского возраста старше 6 лет с бляшечным псориазом (см. таблицы 13 и 15).
Тяжелые формы бляшечного псориаза
Безопасность и эффективность секукинумаба оценивалась в рандомизированном двойном слепом плацебо- и этанерцептконтролируемом исследовании фазы III у пациентов детского возраста от 6 до < 18 лет с тяжелыми формами бляшечного псориаза, определяемыми по индексу площади и тяжести поражения псориазом ≥ 20, 4 балла по системе глобальной оценки исследователя IGA 2011 и ППТ ≥ 10%, которым назначалась системная терапия. Приблизительно 43 % пациентов ранее получали фототерапию, 53% — стандартную системную терапию, 3% — биологические препараты, и 9% имели сопутствующий псориатический артрит.
В исследовании 1 пациентов детского возраста с псориазом оценивались 162 пациента, которые были рандомизированы для получения секукинумаба в низких дозах (75 мг для массы тела < 50 кг или 150 мг для массы тела ≥ 50 кг), секукинумаба в высоких дозах (75 мг для массы тела < 25 кг, 150 мг для массы тела от ≥ 25 кг до < 50 кг или 300 мг для массы тела ≥ 50 кг) или плацебо на 0-й, 1-й, 2-й, 3-й и 4-й неделе с такой же дозой каждые следующие 4 недели или этанерцепта. Пациенты, рандомизированные в группу этанерцепта, еженедельно получали 0,8 мг/кг (до максимальной дозы 50 мг). Распределение пациентов по весу и возрасту при рандомизации описано в таблице 13.
Таблица 13. Распределение пациентов детского возраста с псориазом по весу и возрасту в исследовании 1

Пациенты, рандомизированные для получения плацебо, у которых отсутствовал ответ на 12-й неделе, были переведены в группу секукинумаба низкой или высокой дозы (доза основана на группе согласно массе тела) и получали исследуемый препарат на 12-й, 13-й, 14-й и 15-й неделе, а затем ту же дозу каждый месяц, начиная с 16-й недели. Комбинированные основные конечные точки отображали долю пациентов, достигших индекса площади и тяжести псориаза 75, и «чистого» или «почти чистого» ответа (0 или 1) по системе глобальной оценки исследователя 2011 на 12-й неделе.
Эффективность низкой и высокой дозы секукинумаба была сопоставима с комбинированными основными конечными точками в течение 12-недельного плацебо-контролируемого периода исследования.
Оценки отношения шансов в пользу обеих доз секукинумаба были статистически значимыми для ответов 0 или 1 как по индексу площади и тяжести псориаза 75, так и по системе глобальной оценки исследователя IGA 2011.
Наблюдение пациентов для оценки эффективности и безопасности проводилось до 52 недель после первого введения исследуемого препарата. Доля пациентов, достигших индекса площади и тяжести псориаза 75, и «чистого» или «почти чистого» ответа (0 или 1) по системе глобальной оценки исследователя 2011, показала различия между группами лечения секукинумабом и плацебо при первом посещении после исходного обследования на 4-й неделе, а к 12-й неделе разница стала более заметной. Ответ сохранялся в течение 52 недель (см. таблицу 13). Улучшение показателей индекса площади и тяжести псориаза 50, 90, 100 и 0 или 1 балл по детскому дерматологическому индексу качества жизни (ДДИКЖ) также сохранялись на протяжении 52 недель.
Кроме того, показатели индекса площади и тяжести псориаза 75, 90 и 0 или 1 балл по системе глобальной оценки исследователя на 12-й и 52-й неделе для групп обеих, низкой и высокой, доз секукинумаба были выше, чем аналогичные показатели у пациентов, получавших этанерцепт (см. таблицу 14).
После 12 недель эффективность как низкой, так и высокой дозы секукинумаба была сопоставима между собой, хотя эффективность высокой дозы была выше у пациентов с массой тела ≥ 50 кг.
Профили безопасности низкой и высокой дозы были сопоставимы и соответствовали профилю безопасности у взрослых.
Таблица 14. Обзор клинических ответов у пациентов с тяжелыми формами псориаза на 12-й и 52-й неделях (исследование пациентов детского возраста с псориазом 1) *

Большая доля пациентов детского возраста, получавших секукинумаб, сообщила об улучшении связанного со здоровьем качества жизни, оценка 0 или 1 балл по шкале ДДИКЖ в сравнении с плацебо на 12-й неделе (низкая доза 44,7%, высокая доза 50%, плацебо 15%). До 52-й недели включительно обе группы доз секукинумаба были численно выше, чем группа этанерцепта (низкая доза 60,6%, высокая доза 66,7%, этанерцепт 44,4 %).
Умеренные или тяжелые формы бляшечного псориаза
Предполагалось, что секукинумаб будет эффективен для лечения пациентов детского возраста с умеренной формой бляшечного псориаза на основании продемонстрированной эффективности и связи между экспозицией и ответом у взрослых пациентов с умеренными и тяжелыми формами бляшечного псориаза, а также сходства течения заболевания, патофизиологии и лекарственного эффекта препарата у взрослых и детей при одинаковом уровне экспозиции.
Кроме того, безопасность и эффективность секукинумаба оценивались в открытом многоцентровом исследовании фазы III в двух параллельных группах у пациентов детского возраста от 6 до < 18 лет с умеренными или тяжелыми формами бляшечного псориаза, определяемыми по индексу площади и тяжести поражения псориазом ≥ 12,3 балла и более по системе глобальной оценки исследователя IGA 2011 и ППТ ≥ 10 %, которые были кандидатами на системную терапию.
В исследовании 2 пациентов детского возраста с псориазом оценивались 84 пациента, которые были рандомизированы для получения секукинумаба в низких дозах (75 мг для массы тела < 50 кг или 150 мг для массы тела ≥ 50 кг), секукинумаба в высоких дозах (75 мг для массы тела < 25 кг, 150 мг для массы тела от ≥ 25 кг до < 50 кг или 300 мг для массы тела ≥ 50 кг) или плацебо на 0-й, 1-й, 2-й, 3-й и 4-й неделе с такой же дозой каждые следующие 4 недели. Распределение пациентов по весу и возрасту при рандомизации описано в таблице 15.
Таблица 15. Распределение пациентов детского возраста с псориазом по весу и возрасту в исследовании 2

Комбинированные основные конечные точки отображали долю пациентов, достигших индекса площади и тяжести псориаза 75, и «чистого» или «почти чистого» ответа (0 или 1) по системе глобальной оценки исследователя 2011 на 12-й неделе.
Эффективность низкой и высокой дозы секукинумаба была сопоставима; также было показано статистически значимое улучшение по сравнению с приемом плацебо в анамнезе для комбинированных основных конечных точек. Оцениваемая апостериорная вероятность положительного эффекта лечения составила 100 %.
Наблюдение всех пациентов для оценки эффективности проводилось по меньшей мере 52 недели после первого введения исследуемого препарата (см. таблицу 16). Эффективность (определяемая как индекс площади и тяжести псориаза 75 и «чистый» или «почти чистый» [0 или 1] ответ по системе глобальной оценки исследователя IGA 2011) наблюдалась уже при первом посещении после исходного обследования на 2-й неделе, и доля пациентов, достигших индекса площади и тяжести псориаза 75, и «чистого» или «почти чистого» ответа (0 или 1) по системе глобальной оценки исследователя 2011 в течение 24 недель увеличилась и сохранялись до 52 недели. Улучшение показателей индекса площади и тяжести псориаза 90 и 100 также наблюдалось на 12-й неделе и увеличивалось в течение 24 недель и поддерживалась до 52 недели (см. таблицу 16).
После 12 недель эффективность низкой и высокой дозы секукинумаба была сопоставима между собой. Профили безопасности низкой и высокой дозы были сопоставимы и соответствовали профилю безопасности у взрослых.
Таблица 16. Обзор клинических ответов у пациентов с умеренными и тяжелыми формами псориаза на 12-й и 52-й неделе (исследование пациентов детского возраста с псориазом 2) *

Эти результаты в популяции пациентов детского возраста с умеренными и тяжелыми формами бляшечного псориаза подтвердили прогностические предположения, основанные на эффективности и связи между экспозицией и ответом у взрослых пациентов, упомянутых выше.
В группе низкой дозы 50% и 70,7% пациентов достигли 0 или 1 балла по ДДИКЖ на 12-й и 52-й неделе соответственно. В группе высокой дозы 61,9% и 70,3% пациентов достигли 0 или 1 балла по ДДИКЖ на 12-й и 52-й неделе соответственно.
Эффективность и безопасность секукинумаба оценивали у 86 пациентов в рамках двойного слепого, плацебо-контролируемого, управляемого событиями, рандомизированного исследования фазы III, состоящего из 3 частей, у пациентов в возрасте от 2 до 18 лет с активным ЭАА или ЮПА, диагноз у которых был поставлен на основе модифицированных критериев классификации ЮИА Международной лиги ревматологических ассоциаций (ILAR). В исследование входила открытая часть (часть 1), в рамках которой все пациенты получали секукинумаб до 12 недели. Пациенты, у которых был продемонстрирован ответ ЮИА ACR 30 на 12 неделе, перешли в часть 2, двойную слепую фазу, и были рандомизированы в соотношении 1:1 для продолжения лечения секукинумабом или для начала лечения плацебо (рандомизированная отмена) до 104 недели или до возникновения обострения. Затем пациенты с обострением начали получать лечение секукинумабом в открытом режиме, которое продолжалось до 104 недели (часть 3).
На момент включения в исследование у пациентов было зарегистрировано следующее распределение ЮИА по типам: ЭАА — 60,5% и ЮПА — 39,5 % субъектов, у которых либо был получен неадекватный ответ, либо наблюдалась непереносимость ≥ 1 болезнь-модифицирующего антиревматического препарата (БМАРП) и ≥ 1 нестероидного противовоспалительного препарата (НПВП). На исходном уровне было зарегистрировано применение MTX у 65,1% пациентов; (63,5% [33/52] пациентов с ЭАА и 67,6% [23/34] пациентов с ЮПА). Параллельное лечение сульфасалазином получали 12 из 52 пациентов с ЭАА (23,1%). Пациентам с массой тела < 50 кг на исходном уровне (n=30) вводили дозу 75 мг, а пациентам с массой тела ≥ 50 кг (n = 56) — 150 мг. Возраст на исходном уровне варьировался от 2 до 17 лет: возраст 3 пациентов находился в промежутке от 2 до 6 лет, 22 пациентов — от 6 до 12 лет и 61 пациента — от 12 до 18 лет.
Индекс активности заболевания, ювенильного артрита, (JADAS)-27 на исходном уровне составлял 15,1 (СО: 7,1).
Первичной конечной точкой в периоде рандомизированной отмены было время до обострения (часть 2). Обострение заболевания определялось как ухудшение на ≥ 30% значений по меньшей мере трех из шести критериев ответа ЮИА ACR и улучшение на ≥ 30% значений не более чем по одному из шести критериев ответа ЮИА ACR, а также наличие активной фазы с поражением по меньшей мере двух суставов.
В конце части 1 у 75 из 86 (87,2%) пациентов был продемонстрирован ответ ЮИА ACR 30; они перешли в часть 2.
Первичная конечная точка исследования была достигнута за счет регистрации статистически значимого увеличения времени до обострения заболевания у пациентов, получавших секукинумаб, по сравнению с группой плацебо в части 2. Риск обострения снизился на 72% у пациентов из группы секукинумаба по сравнению с пациентами, получавшими плацебо, в части 2 (отношение рисков = 0,28, 95% ДИ: от 0,13 до 0,63, p<0,001) (рисунок 4 и таблица 17). В ходе части 2 в общей сложности у 21 пациента из группы плацебо произошло обострение (11 человек с ЮПА и 10 человек с ЭАА) по сравнению с 10 пациентами в группе секукинумаба (4 человека с ЮПА и 6 человек с ЭАА).
Рисунок 4. Оценки по методу Каплана — Майера времени до обострения в ходе части 2 

Таблица 17. Анализ выживаемости по времени до обострения заболевания — часть 2

В открытой части 1 все пациенты получали секукинумаб до 12 недели.
На 12 неделе у 83,7%, 67,4% и 38,4% детей были зарегистрированы ответы ЮИА ACR 50, 70 и 90 соответственно (рисунок 5). Начало действия секукинумаба проявилось уже на 1 неделе. На 12 неделе индекс JADAS-27 составил 4,64 (СО: 4,73), а среднее снижение JADAS-27 по сравнению с исходным уровнем — –10,487 (СО: 7,23).
Рисунок 5. Ответ ЮИА ACR 30/50/70/90 пациентов до 12 недели в части 1 *  * Условная оценка отсутствия ответа у пациента, используемая в отношении недостающих значений.
* Условная оценка отсутствия ответа у пациента, используемая в отношении недостающих значений.
Данные в возрастной группе от 2 до 6 лет не позволили сделать определенное заключение из-за небольшого числа пациентов младше 6 лет, включенных в исследование.
Европейское агентство лекарственных средств отказалось от обязательства представлять результаты исследований для препарата Козэнтикс в лечении псориаза у детей в возрасте от рождения до менее 6 лет, а также в лечении хронического идиопатического артрита у детей в возрасте от рождения до менее 2 лет (см. раздел 4.2 для получения информации об использовании у детей).
Большинство фармакокинетических свойств, наблюдаемых у пациентов с бляшечным псориазом, псориатическим артритом и анкилозирующим спондилитом, были схожи.
Абсорбция
После однократного подкожного введения дозы 300 мг в виде раствора у здоровых добровольцев, максимальная концентрация секукинумаба в сыворотке крови составляла 43,2 ± 10,4 мкг/мл через 2-14 дней после введения дозы.
На основании популяционного фармакокинетического анализа, после однократного подкожного введения препарата в дозе 150 мг или 300 мг у пациентов с бляшечным псориазом, максимальная концентрация секукинумаба в сыворотке крови составляла 13,7±4,8 мкг/мл или 27,3±9,5 мкг/мл, соответственно, через 5-6 дней после введения дозы.
После первоначального еженедельного введения в течение первого месяца время достижения максимальной концентрации составило от 31 до 34 дней, базируясь на данных фармакокинетических исследований популяции.
На основе смоделированных данных, максимальные концентрации в равновесном состоянии (Cmax,ss) после подкожного введения 150 мг или 300 мг составили 27,6 мкг/мл и 55,2 мкг/мл, соответственно.
Фармакокинетические исследования популяции показывают, что равновесное состояние достигается через 20 недель при ежемесячном режиме дозирования.
По сравнению с экспозицией после однократной дозы популяционный фармакокинетический анализ показал 2-кратное увеличение максимальной концентрации в сыворотке и площади под кривой (AUC) после многократного ежемесячного введения во время поддерживающей терапии.
Фармакокинетические исследования популяции показали, что секукинумаб абсорбируется у пациентов с псориазом со средней абсолютной биодоступностью на 73%. В исследованиях рассчитанная абсолютная биодоступность находилась в диапазоне от 60 до 77%. Биодоступность секукинумаба у пациентов с ПсА составила 85% на основе популяционной фармакокинетической модели.
Распределение
Средний объем распределения при терминальной фазе (Vz) после однократного внутривенного введения колебался от 7,10 до 8,60 литров у пациентов с псориазом, предполагая, что секукинумаб подвергается ограниченному распространению в периферических отделах.
Биотрансформация
Выведение иммуноглобулина G осуществляется в основном путем внутриклеточного катаболизма после пиноцитоза или рецепторноопосредованного эндоцитоза.
Элиминация
Средний общий клиренс (СОК) после однократного внутривенного введения пациентам с бляшечным псориазом варьировал от 0,13 до 0,36 л/день. В популяционном фармакокинетическом исследовании средний общий клиренс (СОК) составил 0,19 л/день для пациентов с бляшечным псориазом. Пол пациентов не оказывал влияния на СОК. Клиренс не зависел от времени и дозы.
Средний период полувыведения составил 27 дней у пациентов с бляшечным псориазом и варьировал в пределах от 18 до 46 дней после внутривенного введения препарата по результатам исследований.
Линейность (нелинейность)
Фармакокинетические параметры при однократном и многократном введении секукинумаба у пациентов с бляшечным псориазом были определены в нескольких исследованиях с внутривенным введением доз в пределах от 1x0,3 мг/кг до 3x10 мг/ кг и с подкожным введением доз в пределах от 1x25 мг до многократной дозы 300 мг. Экспозиция была пропорциональна дозе во всех режимах дозирования.
Основываясь на результатах фармакокинетического анализа популяции с ограниченным числом пациентов пожилого возраста (n=71 для возраста ≥65 лет и n=7 для возраста ≥75 лет), клиренс у пожилых пациентов и больных моложе 65 лет был схожим.
Пациенты с почечной или печеночной недостаточностью
Фармакокинетические данные для больных с почечной или печеночной недостаточностью отсутствуют. Почечное выведение интактного секукинумаба в виде моноклонального антитела IgG, как ожидается, будет низким и малозначимым. IgG в основном выводится посредством катаболизма, а печеночная недостаточность, как ожидается, не повлияет на клиренс секукинумаба.
Клиренс и объем распределения секукинумаба повышается при увеличении массы тела.
В пуле двух исследований пациентов детского возраста с умеренными и тяжелыми формами бляшечного псориаза (от 6 до менее чем 18 лет) вводили секукинумаб в соответствии с рекомендуемым режимом дозирования для детей. На 24-й неделе у пациентов с массой тела ≥ 25 и < 50 кг среднее значение минимальной концентрации в равновесном состоянии ± СО составляло 19,8 ± 6,96 мкг/мл (n=24) после приема 75 мг секукинумаба, а у пациентов с массой тела ≥ 50 кг среднее значение минимальной концентрации ± СО составляло 27,3 ± 10,1 мкг/мл (n=36) после приема 150 мг секукинумаба. Среднее значение минимальной концентрации в равновесном состоянии ± СО у пациентов с массой тела < 25 кг (n=8) на 24-й неделе составляло 32,6 ± 10,8 мкг/мл после приема 75 мг препарата.
Ювенильный идиопатический артрит
В исследовании у детей пациентам с ЭАА и ЮПА (в возрасте от 2 до менее 18 лет) вводили секукинумаб в соответствии с рекомендуемым режимом дозирования для детей. На 24 неделе у пациентов с массой тела < 50 кг и массой тела ≥ 50 кг среднее значение минимальной концентрации в равновесном состоянии ± СО составляло 25,2 ± 5,45 мкг/мл (n=10) и 27,9 ± 9,57 мкг/мл (n=19) соответственно.
Доклинические данные не выявили особой опасности для человека (взрослых или детей) на основе исследований фармакологической безопасности, повторяющихся доз, исследований репродуктивной токсичности или перекрестной реактивности тканей. Исследования на животных по оценке канцерогенного потенциала секукинумаба не проводились.
6. Фармацевтические свойства
Трегалозы дигидрат
L-гистидин/гистидина гидрохлорида моногидрат
L-метионин
Полисорбат 80 с низким содержанием пероксидов
Вода для инъекций
В отсутствие исследований совместимости не рекомендуется смешивать указанное лекарственное средство с другими медицинскими продуктами.
2 года
Не применять по истечении срока годности.
В защищенном от света месте при температуре от 2ºС до 8ºС.
Не замораживать. Хранить в оригинальной упаковке.
Хранить в недоступном для детей месте!
По 1 мл в стеклянный шприц с закрепленной иглой с защитным колпачком (предварительно заполненный шприц).
По 1 предварительно заполненному шприцу с устройством для пассивной защиты иглы в упаковке контурной ячейковой, заклеенной этикеткой, вместе с инструкцией по медицинскому применению на казахском и русском языках помещают в пачку картонную с контролем первичного вскрытия.
По 1 предварительно заполненному шприцу в автоинжекторе (ручке) с контролем первичного вскрытия, вместе с инструкцией по медицинскому применению на казахском и русском языках помещают в пачку картонную с контролем первичного вскрытия.
Козэнтикс 150 мг, раствор для инъекций, поставляется в одноразовом, предварительно заполненном шприце или предварительно заполненном шприце в автоинжекторе (ручке), предназначенном для индивидуального использования. Шприц или автоинжектор (ручка) должен быть извлечен из холодильника за 20 минут до инъекции, чтобы вещество достигло комнатной температуры.
Перед использованием рекомендуется визуальный осмотр предварительно заполненного шприца. Жидкость должна быть прозрачной. Ее цвет может варьировать от бесцветного до светложелтого. Наличие небольшого пузырька воздуха является нормой.
Не используйте препарат, если жидкость содержит видимые частицы, помутнела или имеет выраженный коричневый оттенок.
Подробные инструкции по использованию приведены в листкевкладыше.
Любой неиспользованный продукт или отходы должны быть утилизированы в соответствии с локальными требованиями.
По рецепту.
7. Держатель регистрационного удостоверения
Новартис Оверсиз Инвестментс АГ
Лихтштрассе 35, 4056 Базель, Швейцария
тел.: +7 (727) 258-24-47
Претензии потребителей направлять по адресу:
Республика Казахстан
Филиал Компании «Новартис Фарма Сервисэз АГ» в Республике Казахстан
050022 г. Алматы, ул. Курмангазы, 95
тел.: +7 (727) 258-24-47
8. Номера регистрационных удостоверений
РК-ЛС-5№024318
9. Дата первичной регистрации (подтверждения регистрации, перерегистрации)
Дата первой регистрации: 01 октября 2019 г.
10. Дата пересмотра текста
Общая характеристика лекарственного препарата доступна на официальном сайте http://www.ndda.kz